Functionless DNA changes more rapidly, functional DNA more slowly. This is one of the fundamental principles of comparative genomics. It’s why people look at the ratio of synonymous nucleotide changes to nonsynonymous nucleotide changes within the coding sequence of genes. It’s why the exons of two related genes will still have strikingly similar sequences after the sequence of the introns have diverged to the point where it’s impossible to even detect homology. It’s also a way to identify which parts of the noncoding sequence surrounding a set of exons are functionally constrained. The bits of noncoding sequence that determine where, and when, and how much, a gene is expressed are by definition, functional, and should diverge more slowly between even related species than the big soup of functionless noncoding sequence that the functional bits of a genome float in. These conserved, functional, noncoding sequences are called, unimaginatively, conserved noncoding sequences (CNS).*
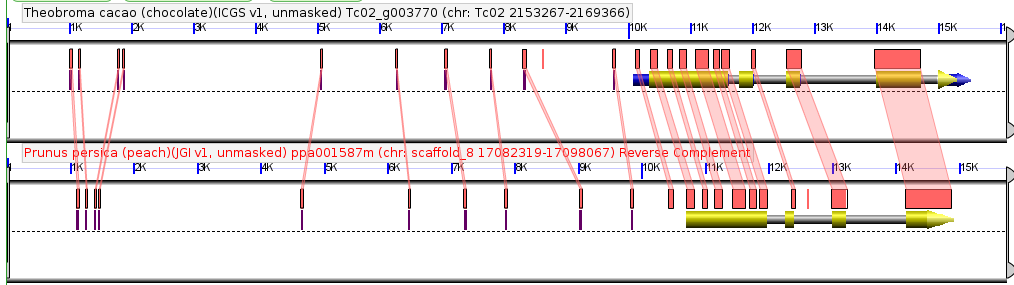
Comparison of a single syntenic orthologous gene pair in the genomes of peach and chocolate. Coding sequence marked in yellow, introns in gray, annotated UTRs in blue. Red boxes are regions of detectably similar sequence between the same genomic region in these two species. Taken from CoGePedia.
I’ve been playing with CNS since I first opened a command line window back as a first year grad student. The smallest CNS we’d consider “real” were 15 base pair exact matches between the same gene in two species. On the one hand, this seemed a bit too big, because I know lots of transcription factors bound to motifs as short as 6-10 base pairs long. On the other hand this seemed a bit too short because I’d see 15 base pair exact matches that couldn’t be real a bit too often (for example a match between a sequence in the intron of one gene, and the sequence after the 3′ UTR of another).
15 bp represented a compromise between the two concerns pushing in opposite directions. Then, in the fall of 2014, a computer science PhD student walked into my office and asked if I had any interesting bioinformatics problems he could work on. The result was a new algorithm (STAG-CNS) which was both more stringent at identifying conserved noncoding sequences and able identify shorter conserved sequences than was previously possible. It achieved both of these goals through the expedient of throwing genomes from more and more species at the problem.