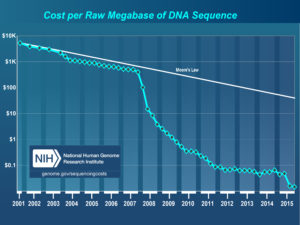
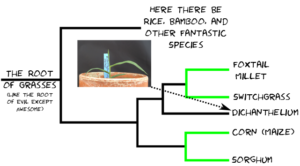
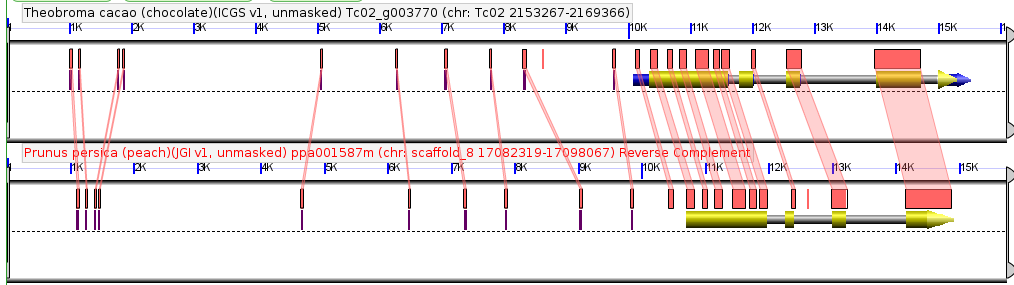
A paper eight years in the making and sixteen months in review. A real credit to Guangchao. I don’t think it ever would have come out without his dedication above and beyond what anyone could expect from a postdoc – James.
A team of researchers, led by Dr. Guangchao Sun and Prof. James Schnable of the University of Nebraska just published the genome of paspalum (Paspalum vaginatum) alongside evidence that paspalum may employ some tricks that could help its relative, corn, grow better with less fertilizer.

Center greenhouses.
The Nebraska team, in collaboration with researchers from the Department of Energy’s Joint Genome Institute, the University of Georgia, and the HudsonAlpha Institute for Biotechnology, wanted to use the new genome sequenced this resilient grass to understand what makes the grass so much more stress tolerant that closely related crops, including corn and sorghum.
Using comparative transcriptomic and metabolomic analyses of paspalum, corn, and sorghum under optimal and stress conditions they identified a specific metabolic pathway — trehalose — that was being produced in paspalum, but not in corn and sorghum, in response to stress.
The team used a strategy called chemical genetics to convince corn plants to also start producing and accumulating trehalose and showed that these corn plants grew faster and larger in conditions without enough fertilizer than corn plants without extra trehalose. Finally, the team used a combination of experiments to show that the reason these corn plants were able to grow more with less fertilizer was because of a process called autophagy, essentially a recycling program within plant cells that breaks down old, damaged, and unneeded proteins into spare parts that can be used to make new proteins.

plants grown without enough nitrogen fertilizer.
“I’m so excited to see this story come out,” said Prof. Schnable, who is currently taking leave from the University of Nebraska while working at Google. “Paspalum is vegetatively propagated which means we cannot just save seeds, we always have to keep living plants for our research. There was a period where no one remembered to water the paspalum plant for a couple of months. But the plant was completely fine. In fact it usually grows so fast it’ll try to invade the pots of neighboring plants and the greenhouse manager has to yell at me or folks in my lab to come down and trim it. With this genome sequence and all the great work Guangchao Sun and the team have done, we finally are starting to understand just what makes this plant so
resilient.”
Sun, G., Wase, N., Shu, S. et al. Genome of Paspalum vaginatum and the role of trehalose mediated autophagy in increasing maize biomass. Nature Communications 13, 773 (2022) doi: 10.1038/s41467-022-35507-8